Optimized analytical workflow for single-nucleus transcriptomics in main metabolic tissues
2024-11-21
1 Introduction
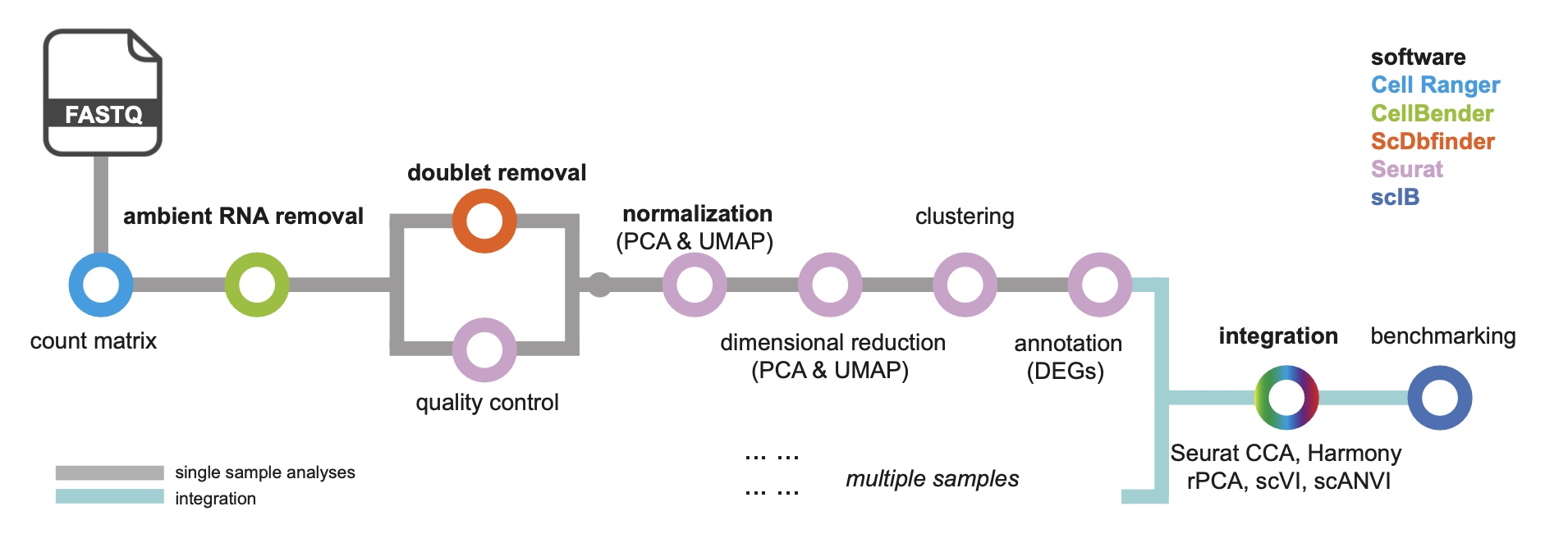
This is a benchmarked and optimized workflow for analyzing single-nuclei RNA sequencing (snRNA-seq), tailored to the analysis of metabolically active tissues. This workflow includes key steps from preprocessing to downstream analyses. This technical note aims to serve as a standard protocol for researchers seeking to apply snRNA-seq to metabolic tissues including adipose tissue, muscle, hypothalamus and liver to uncover novel regulatory mechanisms, identify new therapeutic targets, and ultimately contribute to the development of more effective treatments for metabolic disorders.
This workflow encompasses a series of procedures, including the alignment and quantification of raw FASTQ files, followed by the recommended removal of ambient RNA after generating the raw matrix. Additionally, it involves the potential identification and removal of doublets using scDblFinder, normalization of gene expression values, multiple approaches for data integration, and comprehensive benchmarking steps to ensure robust results.
 Citation: Dong et al, An optimized practice for upstream analysis of single nucleus transcriptomics in key metabolic tissues
Citation: Dong et al, An optimized practice for upstream analysis of single nucleus transcriptomics in key metabolic tissues